
- Unstained cells at a concentration of 3 – 5 million cells/mL for 10 – 20 measurements.
- A spreadsheet with the extracted flow cytometer baseline data for the current bead lot with an additional column for 2.5 robust standard deviation (rSD).
- Retrieve the baseline data from BD Cytometer Setup and Tracking (CST) software as follows:
- CYTOMETER MENU → CST → Find the Baseline for the current bead lot → Right click on the file name and select Export.
- Relabel the file with your name and experiment e.g. [insert your name]_[experiment]_Panel [date]
Table 1: Example Baseline Data for BD Cytometer Setup and Tracking Beads

The minimum PMT voltage setting for each detector is determined so that the resulting rSD of the unstained cells is approximately 2.5x the rSD of the electronic noise (2.5 rSDen).
Purpose: to ensure that electronic noise does not interfere with measurements with dim fluorescence and/or negative events and that you are aware of these limits should it be necessary to decrease the voltages lower than these values in Rule 2 (below).
The Short Way:
- Use the application settings features of BD FACS DIVA (grey plot regions) to help place your negative cells at 2.5x rSDen.
- Refer to your training notes for instructions on how to set this up (University of Toronto Flow Cytometry Facility Core Clients Only)
The Long Way:
- Create a plot of FSC and SSC and gate the cells of interest.
- Create a Statistics Box and select the FSC x SSC population (P1) and the rSD values from the statistics tab for every parameter.
- Sequentially record data files of at least 5000 events starting from the CST voltages, reducing the voltage in increments of 25 until the rSD of the unstained cells comes into line with the values calculated in the last column of the baseline report spreadsheet (refer to Table 1’s example).
- Make a note of these voltages on a worksheet – these are the minimum voltages that may be used to ensure that no more than 10–20% of electronic noise contributes to the biological background of the sample.
- Data from a statistics box can be exported to an Excel worksheet with the desired information.
- The minimum acceptable voltage can be extrapolated by plotting rSDen voltage on a graph.
The Short Way:
- Acquire data for a fully stained tube of cells and reduce the voltages for any detectors where data is off scale positively.
- Ensure to leave enough room at the top of the FSC vs SSC plot so that unexpectedly bright positives can still be captured on scale.
The Long Way:
- Refer to the Linearity Max Channel column of the extracted baseline report; the median fluorescence intensity (MFI) of your fluorochrome should not exceed this value.
- Create a statistics box with the MEDIANS for each fluorescent parameter and acquire data for cells stained with one color only (or with colors that are well-separated and have no spectral overlap).
- Note: The measured fluorescence must be biologically relevant therefore beads cannot be used in this process.
- If there are populations with a larger spread (wide %CV’s) decrease the voltage so that the brightest events remain within the linear range of the detector.
- There should always be some extra room at the top of the scale to accommodate events that have unexpectedly high fluorescence.
- Compensation controls should also fall within the linear range of the detector.
- If the compensation controls are too bright, it is best to remake the samples with lower antibody concentrations as opposed to decreasing voltages to accommodate the bead fluorescence.
If the positives are so far up the scale using the minimum acceptable voltage that they are in the nonlinear range or going off scale, the voltage must be lowered to properly view the positive data regardless of where the negatives reside.
Placing negative cells at 2.5x rSDen does not always result in the best resolution between the negative and positive fluorescence. For example, at the minimum voltage, the separation between negative and positives can be reduced when compared to voltages closer to the CST values; this can consequently have a negative impact on the detection of weakly expressed antigens. Alternatively, when too much voltage is applied, negative populations can spread, especially in channels where there is little to no detectable autofluorescence (red emissions), therefore it is important to optimize the resolution of all stains, paying particular attention to dim fluorochromes and weakly expressed markers, to ensure their correct identification by characterizing the Stain Index over a wider range of voltages.
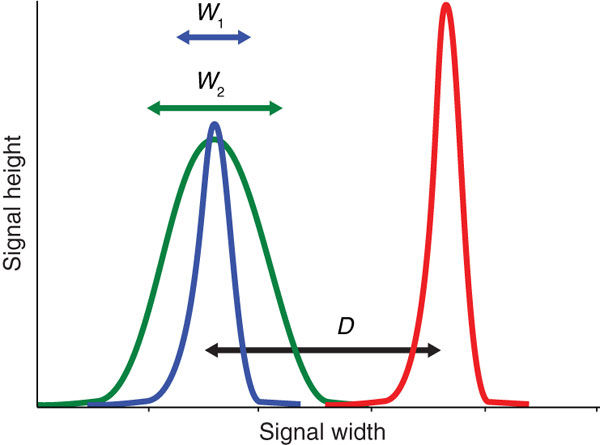
Figure 1: The effective brightness of a reagent depends on the difference (D) between the positive (red) and the negative (blue, green) populations and the spread of the negative population (W). The stain index is a useful metric for normalized signal over background (Maeker & Trotter 2008).
The following method will help to minimize the spread of the negatives while optimizing the resolution of the stains for each detector.
Procedure: Acquire data for the single stained control cells, over a wider range of voltages to determine where the best resolution is obtained. Use this voltage instead of the rSDen as your starting voltage.
- Draw histograms for each parameter and draw positive and negative interval gates on the plots.
- Create a statistics box with the MFIpos, MFIneg and SDneg for each parameter.
- Create a spreadsheet on which you can input data directly from BD FACSDIVA statistics to calculate Stain Index as you change voltages. (you may also export the statistics from BD FACSDIVA and format the resulting table for this calculation).
STAIN INDEX = [MFIPOS – MFINEG]/(2.5x SDNEG)
- For populations that smear (no easily identifiable negative/positive boundary) set some unstained aside so that each time you change the voltage, data is acquired for the negative cells alone, in addition to the stained cells.
- Use the negative cells alone in the stain index calculation (MFIneg).

